Advantages:
-
Novel vector, composition, and method of treating a neurological disorder with UBE3A deficiency.
-
High fidelity to human Angelman syndrome.
-
Facilitates comprehensive symptom representation for diverse treatment investigations.
-
Allows innovative experiments for pioneering treatment development.
Summary:
Angelman syndrome (AS) is a rare and complex neurological disorder triggered by disruptions in the maternally inherited allele encoding the UBE3A gene, which encodes E6-AP, a ubiquitin ligase. This leads to a neuronal-specific imprinting and transcriptional silencing of the paternal allele, resulting in a predominant maternal expression of UBE3A within the brain and giving rise to the clinical manifestations of AS. The AS rat model, created through full Ube3a gene deletion using CRISPR technology, exhibits a spectrum of deficits reminiscent of human AS symptoms. Disturbances in motor coordination, sociability, and memory, coupled with EEG anomalies, render the AS rat a close representative of the human condition.
Research in the AS model has revealed synaptic plasticity deficiencies, notably in long-term potentiation (LTP) and long-term depression (LTD). USF researchers demonstrated that exogenous E6AP protein supplementation therapy was able to rescue LTP deficits, offering a potential therapeutic approach. Moreover, the investigators have developed a novel vector and UBE3A protein construct with additional sequences that allow the secretion from cells and the uptake by neighboring cells. Treatment with this vector demonstrated significant potential in mitigating cognitive and motor impairments. In summary, the novel UBE3A vector can be used for gene therapy to confer a functional E6-AP protein into the neurons and rescue disease pathology.
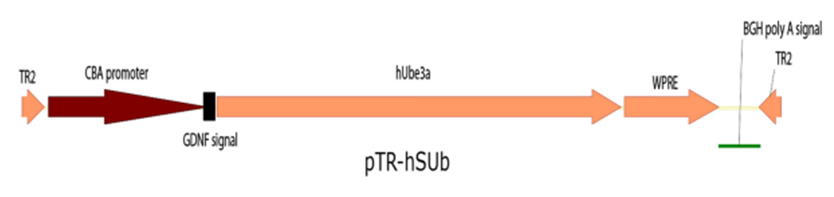
Depicts the human UBE3A gene engineered for secretion within an adeno-associated viral vector, incorporating a signal sequence derived from GDNF.
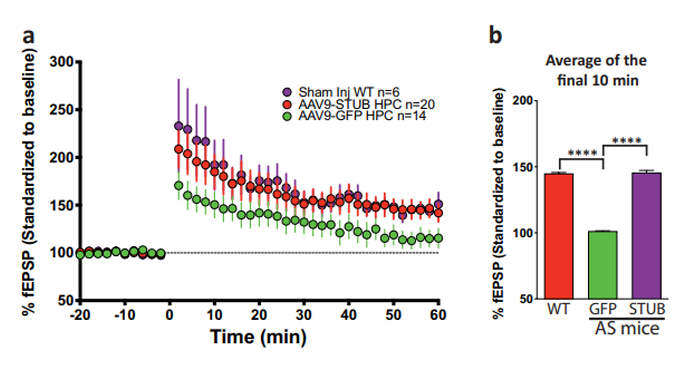
STUB retains function in treated AS mice.
Desired Partnerships:
- License
- Sponsored Research
- Co-Development